![FAIRtracks-logo-light-white-320-[fixed].png](/_nuxt/img/6639f7d.png)
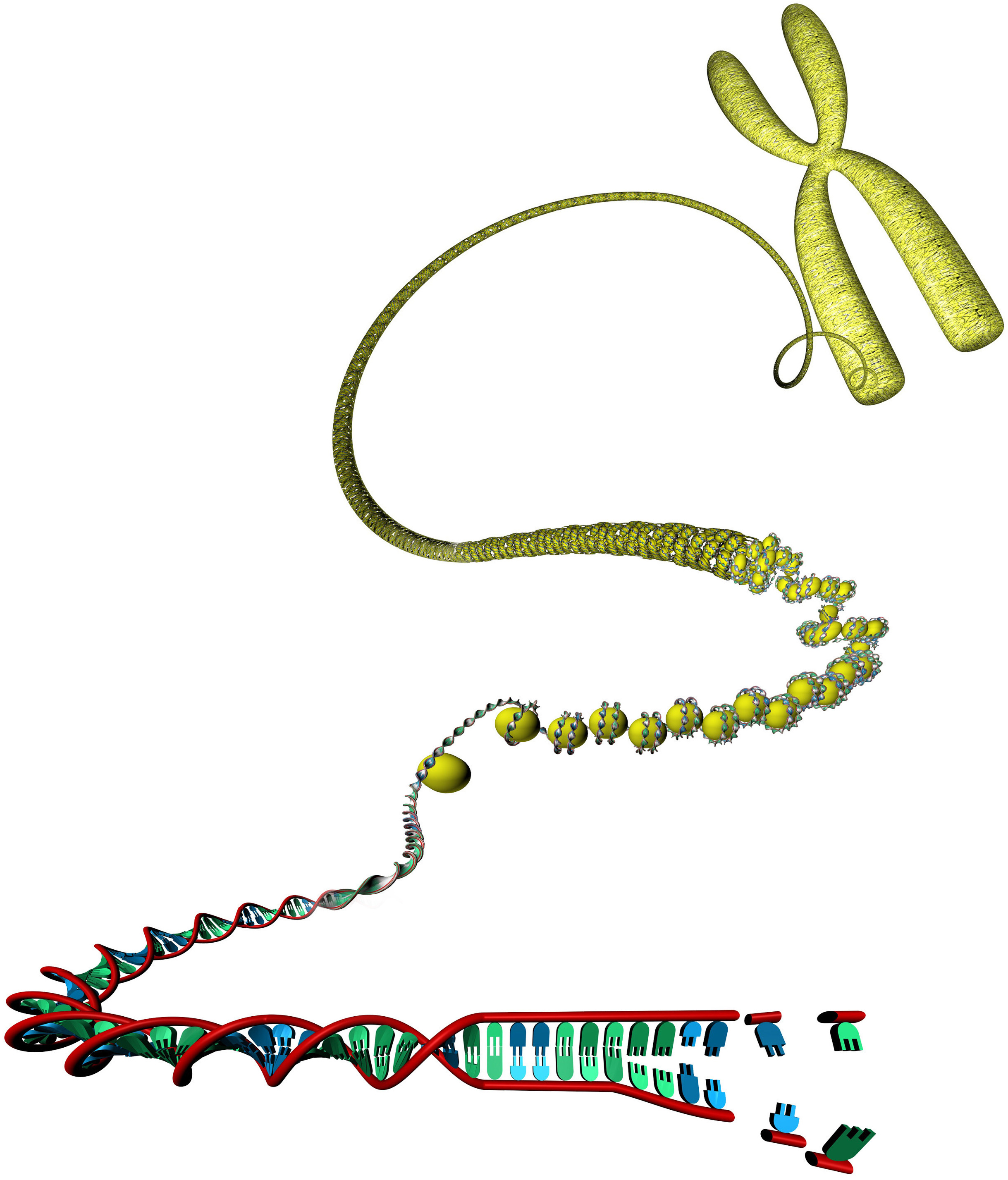
Figure 1.1: Unwinding of a DNA molecule from its compact chromatin form. Each nucleotide pair represents a coordinate position which can be annotated with genomic track data. [Image is © scienceDISPLAY – stock.adobe.com]
Genomic Tracks
Tracks annotate the DNA with condensed data
Genomic tracks refer to data files that annotate DNA reference sequence positions and can be visualized in genome browsers. Track files represent summaries of the raw data according to specific criteria and granularity. For example: “hot spot” regions (with a high number of reads), values deviating from expectations, or cross-genomic links representing closeness in 3D. In essence, the condensed data in track files relate to the raw data much like an abstract describes a scientific publication. This data reduction allows researchers to scan large amounts of data to define a hypothesis before carrying out more accurate analyses.
A genomic track relates to the raw data much like an abstract describes a scientific publication.
Types of data. Many types of data from genomic analyses can be represented as genomic tracks, for example:
- Gene regions, repeating elements, conserved regions
- Chromatin accessibility (e.g., DNase I Hypersensitivity)
- Binding of Transcription Factors (TFs) to DNA
- Histone modifications along the DNA
- Gene expression, Gene fusions, Transcription Start Sites (TSS)
- Cis-regulatory elements (promoters, enhancers, ...)
- DNA methylation
- 3D chromatin interaction
- Single Nucleotide Polymorphisms (SNPs) from Genome-Wide Association Studies (GWAS)
- Single Nucleotide Variants (SNVs) and Copy Number Variants (CNVs) in cancer cells
- Virus insertion sites
- plus many more
De facto standard. Gradually, genomic tracks have also become a de facto standard to store, distribute and analyze genome-wide datasets, mainly because of their efficient compression and indexing utilities. Many bioinformatics analyses are now being distributed, either privately or publicly, using related file formats. Tracks are still predominantly used for graphical display, but can also be queried by statistical analysis tools, such as the GSuite HyperBrowser , EPICO, or DeepBlue.
Figure 1.1: Unwinding of a DNA molecule from its compact chromatin form. Each nucleotide pair represents a coordinate position which can be annotated with genomic track data. [Image is © scienceDISPLAY – stock.adobe.com]
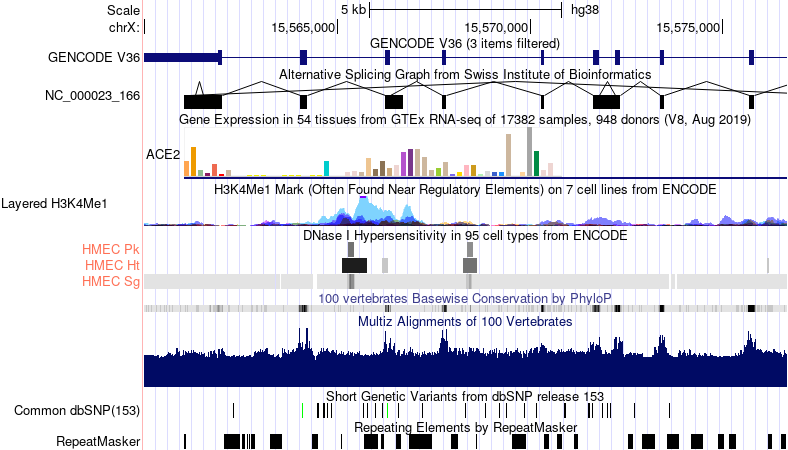
Figure 2.1: Some different types of genomic tracks visualized in the UCSC Genome Browser
Genome Browsers
Genomic tracks are made for genome browsers
Genomic track files were originally designed and optimized to be displayed within genome browsers. A genome browser is a type of genome-wide visualization software able to display various datasets as parallel tracks along the DNA sequence coordinates of typically reference genomes. A multitude of genome browsers are available as online web services on top of a database of track files and/or as installable software. A selection genome browsers are listed in Table 2.1.
The FAIRtracks team advocate the use of the term "track" also in analysis scenarios outside the realm of genome visualisation.
Generality of the term "track". The term "track" implies a mental visual model of elements located along some line. In the domain of genomics this typically relates to a coordinate system defined by a reference genome sequence collection. This is a crucial model to keep in mind also for non-visual analysis scenarios. There exists, to our knowledge, no other term in broad use which is both general and precise enough to cover the same heterogeneity of data content, data representation and analysis scenarios as the term "track" (or a variation thereof). Hence, the FAIRtracks team advocate the use of the term "track" also in analysis scenarios outside the realm of genome visualization.
Figure 2.1: Some different types of genomic tracks visualized in the UCSC Genome Browser
| Genome Browser | Web service | Scientific domain | Installable software | Prog. language | Library |
|---|---|---|---|---|---|
| Ensembl | Yes | General | Yes | Perl / Javascript | No |
| UCSC Genome Browser | Yes | General | Yes | C / HTML | No |
| UCSC Xena | Yes | Cancer multi-omics | Yes | Clojure / Javascript | No |
| WashU Epigenome Browser | Yes | Epigenomics | Yes | Javascript | No |
| Zenbu / FANTOM | Yes | Gene regulation | Yes | C++ / Perl / Javascript | No |
| GTEx Locus Browser | Yes | Human variation (gene-centric) | Only demo | Javascript | No |
Table 2.1: Various genome browsers deployed as web services with possibly domain-restricted track databases and/or available as installable software. Please report any errors or omissions to our GitHub repo as an issue, or provide a PR. For more complete lists of genome browsers, please visit bio.tools or the awesome-genome-visualization web page (genome browser software only).

Figure 3.1: The Experiment Matrix view in the ENCODE data portal, one of the most comprehensive data portals with well-annotated metadata.
Track Collections
Genomic tracks are scattered across collections divided by species, domains, and consortia
Significant investments have gone into the generation of genomic track data both within large consortia and independent groups. However, no central repository exists dedicated to storing track files and curated metadata. When track files are generated in the context of larger consortia, they are typically indexed and made searchable through dedicated data portals. In addition, there exists many specialized "secondary" repositories focused on specific data types or domains. Hence, the existing track repositories are divided by species (mainly human vs. other systems) and/or domain (e.g., epigenomics, cancer, common variants, or rare-disease variants), as listed in Table 3.1.
No central repository exists dedicated to storing track files and curated metadata.
Track hubs. Smaller projects may choose to serve the track files through dedicated "track hubs", which are listed at the Public Hubs page at UCSC or at the EMBL-EBI-hosted Track Hub Registry. However, as serving a track hub requires a bit of maintenance over time, it is not uncommon that they are shut down after some years. Also, the metadata provided with track hubs are often only provided at the track collection level and not at the level of the individual files. In the absence of a data portal or a track hub, the track data is often not indexed other than what is available through the generic interfaces of the main deposition databases (such as GEO or ArrayExpress).
Using tracks for high-level analysis and comparison. In general, data in track files are often heavily influenced by the exact parameters and tools used in the pipeline and can contain biases and artifacts. One common recommendation is thus to reanalyze the raw data in a homogeneous way for reuse in new scientific projects. However, the relative simplicity and accessibility of track data still makes them ideal for higher level comparison and analysis, for instance at an early hypotheses-generating step of a research project, or when there is a need to relate novel findings to existing data to make sense of them.
Cumbersome to reuse tracks in practice. Currently, significant legwork is required in order to identify, collect and consolidate a set of tracks to be used for a given research project. This is not the least due to the fragmented landscape of track repositories and their underlying data models. Moreover, consolidated track analysis is also complicated by low quality metadata annotations, like erroneous and missing metadata, duplicate attributes and/or records. More systematic deficiencies include difficulties accessing metadata and data, and even a lack of adherence to the established formats.
Figure 3.1: The Experiment Matrix view in the ENCODE data portal, one of the most comprehensive data portals with well-annotated metadata.
| Database name | Scientific domain | Organism(s) | Assay type(s) | Organisation(s) | Notes |
|---|---|---|---|---|---|
| Array Express | General | Multiple | Multiple | EMBL-EBI | Data warehouse |
| Gene Expression Omnibus (GEO) | General | Multiple | Multiple | National Center for Biotechnology Information | Data warehouse |
| ENCODE | Epigenetics | H. sapiens, M. musculus | Multiple | ENCODE Project (NIH) | Contains Roadmap and ModENCODE data, data in Array express, GEO and IHEC |
| Roadmap Epigenomics | Epigenetics, Stem cells, primary ex vivo tissues | H. sapiens | Multiple | Roadmap Epigenomics Mapping Consortium (NIH) | |
| FAANG | Epigenetics | B. indictus, B. bubalis, B. taurus, C. hircus, D. labrax, E. caballus, G. gallus, O. mykiss, O. aries, S.salar, S. scrofa | Multiple | Functional Annotation of ANimal Genomes project (FAANG) |
Table 3.1: Various repositories hosting track collections: general repositories, consortia-specific data portals, and secondary repositories focused on particular data types or domains. Please report any errors or omissions to our GitHub repo as an issue, or provide a PR.
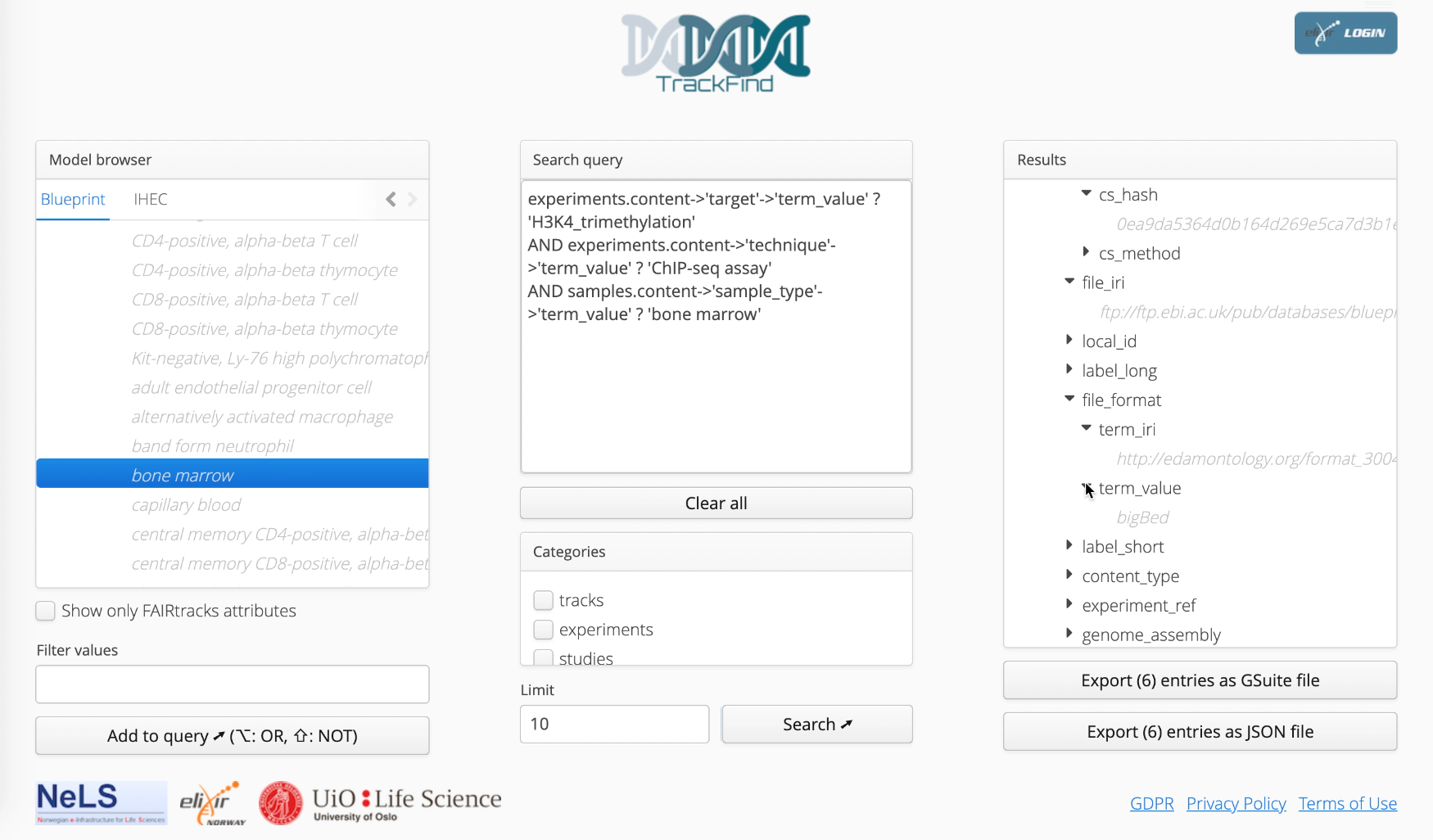
Figure 4.1: TrackFind provides both a web user interface as well a REST API to allow access by downstream tools and scripts. In the web portal, TrackFind provides a categorical browser of the metadata fields defined by the FAIRtracks standard, as well as complete lists of the different values that are present in the indexed metadata for a particular field. Through the graphical user interface, these values can be used to define categorical search queries (in the form of SQL queries). The results of the queries are available for browsing or downloadable as JSON or GSuite formats.
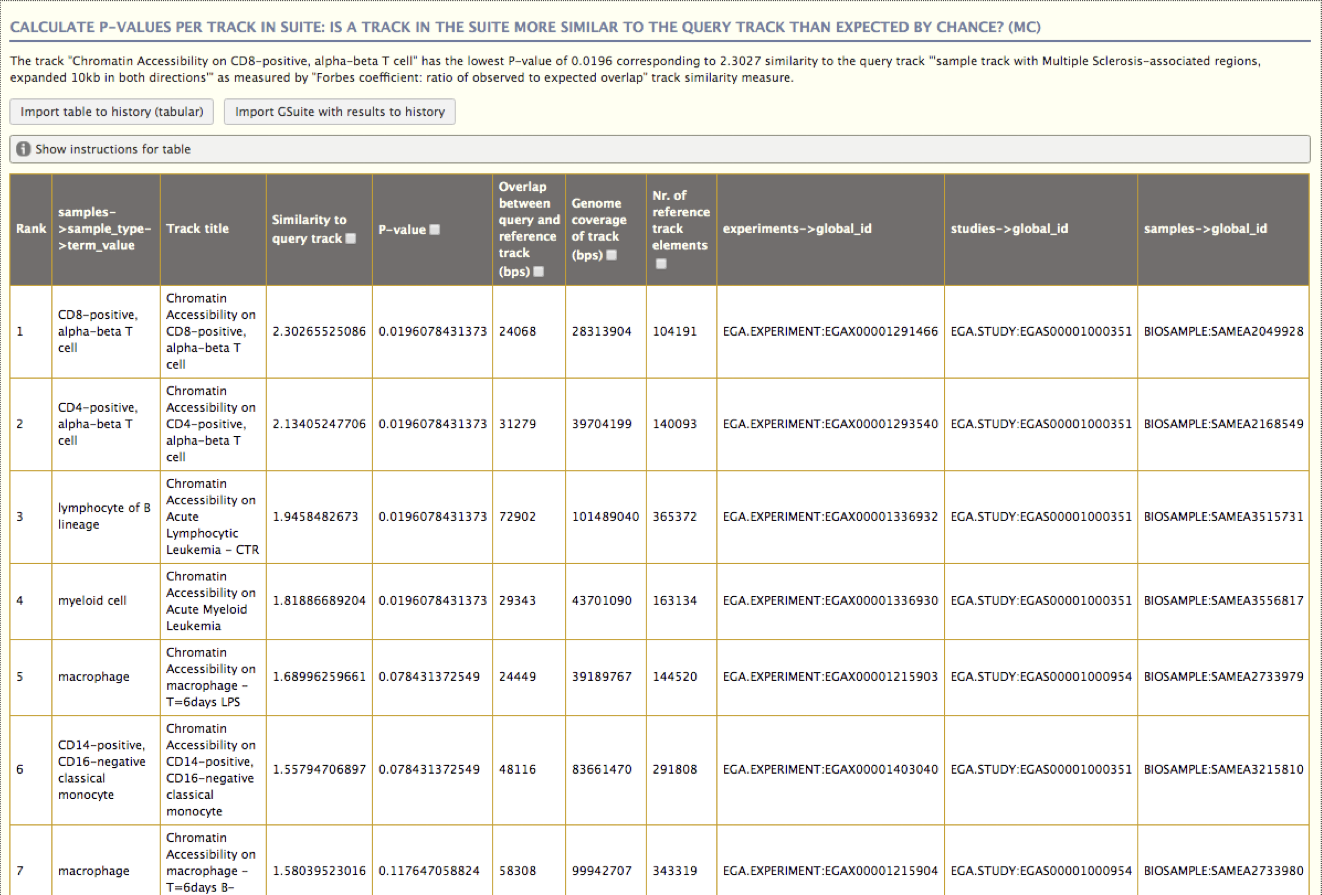
Figure 4.2: An example analysis in the Genomic HyperBrowser, comparing a single set of variants associated with Multiple Sclerosis (MS) with a collection of tracks from BLUEPRINT with sites of open chromatin for various cell types (based on the DNaseI HS assay). This track collection was imported using the integrated TrackFind client in the form of a GSuite metadata file. The HyperBrowser TrackFind client tool illustrates a unconventional approach towards dataset discovery in that the search consists of a series of filtering steps on mostly categorical fields, where the possible values to choose from are dynamically reduced according to what is possible based on the previously selected filtering criteria.
Finding Tracks
FAIRtracks provides discovery of genomic datasets through indexing and search of harmonized track metadata
As introduced above, genomic tracks are condensed data files which are routinely generated through standardized data processing workflows when novel genomic datasets are published. They are typically deposited alongside the raw data files in larger datasets related to scientific studies. Considered through the lens of data discovery, track files can be considered potentially very useful but underutilized, entrypoints into the datasets they are a part of.
Considered through the lens of data discovery, track files can be considered potentially very useful — but underutilized — entrypoints into the datasets they are a part of.
Data-driven discovery. Since track files in themselves contain summary-level data condensing certain aspects of the raw dataset, there is the powerful potential for data-driven approaches for both discovering public data relevant to a particular research scenarios, but also to extract novel knowledge based on the relations between the summary-level track files themselves (see the section on Analyzing tracks, coming soon below). With the recent surge of machine learning technologies based on e.g. deep learning, this potential has not become smaller. However, without precise and uniform annotation of metadata to the track data files, the usefulness of such approaches will be limited. Furthermore, while there exists many rich data portals and other services that support search capabilities (as described above), they are all limited in scope and therefore also in the data models. In addition to annotation of uniform track metadata, there is thus also a need for harmonized search functionality.
![FAIRtracks-logo-transparent-180-[fixed].png](/_nuxt/img/1d83130.png)
The FAIRtracks metadata standard. The FAIRtracks project maintains a draft standard for metadata about genomic tracks. The aim is that the standard will function as a minimal metadata exchange standard for harmonization of heterogeneous track metadata available in various track collections, as described above. Once processes have been set up to transform existing or novel track collection to follow the FAIRtracks standard (see FAIR data and FAIRtracks), the harmonized metadata can be integrated into services for data discovery.
TrackFind and downstream software. As part of the FAIRtracks project, we provide the central service TrackFind for browsing and search in track metadata according to the FAIRtracks standard. A key feature of TrackFind is the ability to integrate search functionality in other software frameworks and tools. Currently, this integration can be implemented through the TrackFind REST API. We aim to develop libraries in common programming languages is that will wrap this API to simplify such integration. Any contributions in this regard is highly appreciated! Currently, we have integrated TrackFind with the GSuite HyperBrowser (see Figure 4.2) and partly in EPICO.
Interoperability of the FAIRtracks standard. TrackFind also illustrates an important aspect of the FAIRtracks metadata standard: as a minimal standard, it is designed to be interoperable with other resources and serviced that contains more detailed content. Hence, TrackFind provides the possibility to launch external websites describing particular vocabulary terms or records detailing source material. For more about this, see sections on Identifiers and Ontologies.
Figure 4.1: TrackFind provides both a web user interface as well a REST API to allow access by downstream tools and scripts. In the web portal, TrackFind provides a categorical browser of the metadata fields defined by the FAIRtracks standard, as well as complete lists of the different values that are present in the indexed metadata for a particular field. Through the graphical user interface, these values can be used to define categorical search queries (in the form of SQL queries). The results of the queries are available for browsing or downloadable as JSON or GSuite formats.
Figure 4.2: An example analysis in the Genomic HyperBrowser, comparing a single set of variants associated with Multiple Sclerosis (MS) with a collection of tracks from BLUEPRINT with sites of open chromatin for various cell types (based on the DNaseI HS assay). This track collection was imported using the integrated TrackFind client in the form of a GSuite metadata file. The HyperBrowser TrackFind client tool illustrates a unconventional approach towards dataset discovery in that the search consists of a series of filtering steps on mostly categorical fields, where the possible values to choose from are dynamically reduced according to what is possible based on the previously selected filtering criteria.
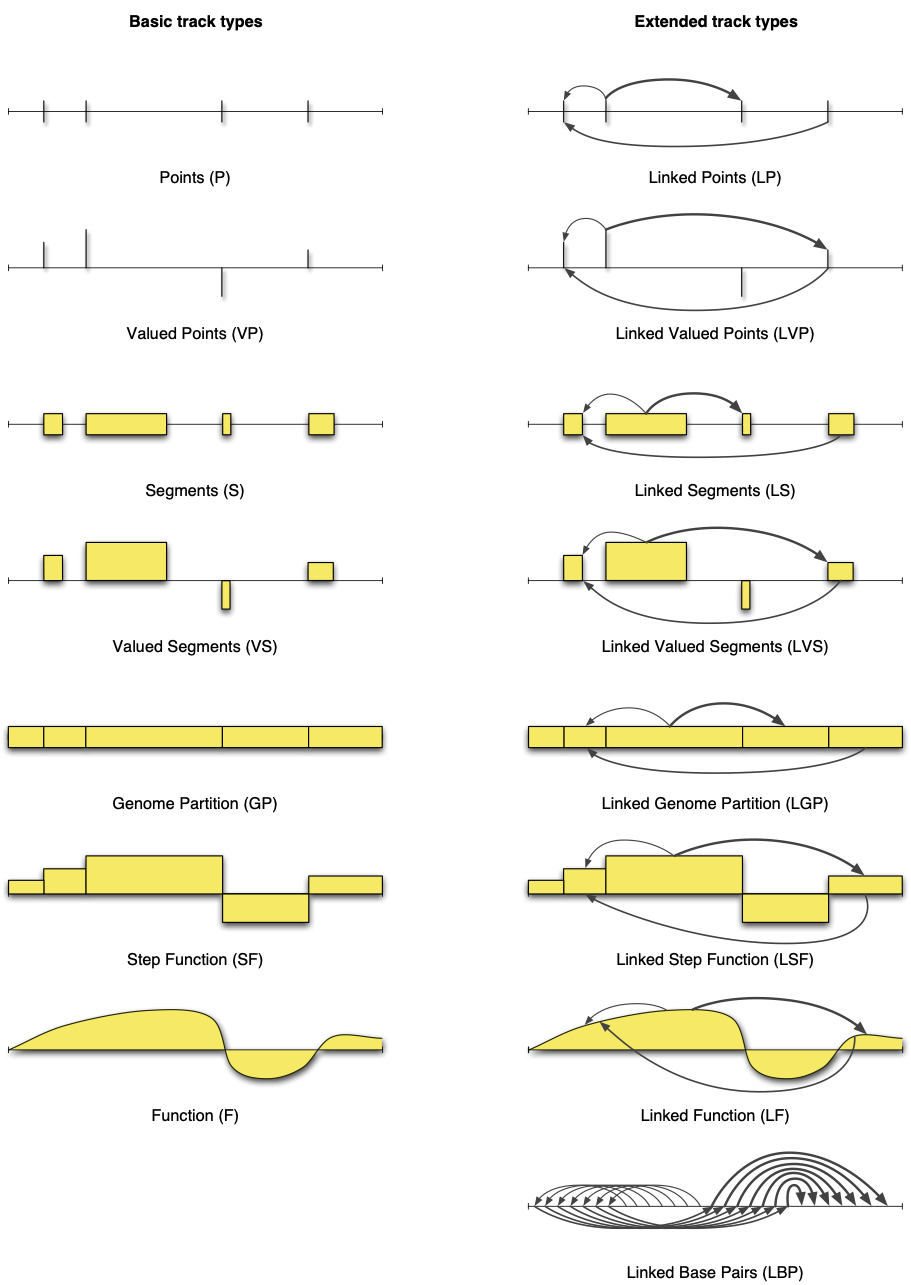
Figure 5.1: Geometrical delineation of genomic tracks into 15 track types [Gundersen, S et al., 2011]
Track Types
Track data can be categorized into 15 theoretical track types
As exemplified in the section Genomic tracks, a multitude of different types of genome-wide data, experimental and otherwise, can be condensed in the form of track files. The variety of data that can be expressed in track form has consequences for the representation of the data, both for mathematical modeling and for the file formats used for storage and exchange.
Track types. Gundersen, S et al. (2011) reviewed and categorized the landscape of track data representation. This paper proposed a unified conceptual framework which delineated the model space into 15 possible mathematical/geometrical models, named track types. These 15 models can be used to represent, visualize, and analyze fundamentally different types of track data.
Gaps, lengths, values, and interconnections. Logically, these track types correspond to whether four different core properties of the data are present: gaps, lengths, values, and interconnections. Figure 3 illustrates the similarities and differences of these 15 different types of track data. If we look closely at the fifteen combinations, starting at the top left of Figure 3 and moving downwards, an interesting pattern appears. From Gundersen, S et al., (2011) 1:
We start at the base case where the only core informational property is the gaps between the track elements. In this case, each track element represents an exact base pair location on the genome, denoting e.g. viral insertion sites. We call this track type Points (P). Adding informative values to this case, e.g. associating SNPs with allele frequencies, we get the track type Valued Points (VP). In the next two cases, the property lengths is added, resulting in the track types Segments (S) and Valued Segments (VS). Segments are probably the most common track type of existing tracks, representing e.g. genes or exons, TF binding sites, or DNA methylation regions. Valued segments could denote e.g. genes with associated expression levels.
Moving on, we remove the values and gaps properties, leaving only lengths. Such tracks consist of segments covering all base pairs of the genome, i.e. a partition of the genome into potentially unequal pieces. Hence, the track type is called Genome Partition (GP). Examples of this track type include the partition of a genome into chromosomes or predicted chromatin states. Adding a value to each part of a partition creates a Step Function (SF), covering the whole genome with values. Examples of such tracks include mapped sequence read counts (e.g. a BAM file), or the signal files that are typically generated in a ChIP-seq workflow. Removing the lengths core property, the step function track is transformed into a track of type Function (F), where every base pair has an associated value. Examples of function tracks are tracks with close dependency on the genome sequence, such as GC content tracks, or the actual genome sequence itself. We call the seven track types outlined here for basic track types.
The fourth core informational property, interconnections, can be envisioned as an orthogonal extension to the previous discussion. Adding interconnections, or edges, to the seven track types previously outlined (first column of Figure 3) defines linked versions of the same track types, e.g. Linked Segments (LS) or Linked Step Function (LSF) (second column of Figure 3). Examples of linked track dataset include ChIA-PET chromatin interactions (**_Linked Segments, LS) and Hi-C contact maps (Linked Genome Partition, LGP**)_.
[...]
To complete the picture, a last track type needs to be defined. If only the interconnections core property is defined, track elements do not have gaps between them, lengths, or values. [...] The linked base pairs track type is mostly suggestive at this point. [...] We refer to the eight linked track types as the extended track types.
Categorizing analyses through track types. The track type concept is useful not only for categorizing tracked data itself but also for a categorizing possible analyses in terms of the track types of their input and output files. For example, ChIP-seq peak calling can be viewed as a data reduction operation, transforming input track files of type Step Function (SF) (i.e. sequence read files) into output track files of type Valued Segments (VS), where the values represent the intensities of the peaks that are called.
Supported by FAIRtracks. Note that all 15 track types are supported by the FAIRtracks metadata standard, as well as the GTrack and GSuite file formats, which are all described in the Standards page.
- The quote is slightly edited. Text formatting have been added and some examples have been substituted with more recent ones.↩
Figure 5.1: Geometrical delineation of genomic tracks into 15 track types [Gundersen, S et al., 2011]